Alport综合征(Alport syndrome,下文简称AS),是2018年被纳入第一批罕见病目录的疾病。据我国资料显示,AS在肾活检患儿中占遗传性肾脏疾病的81.82%,那么究竟什么是Alport综合征呢?
Alport综合征又称眼-耳-肾综合征,是一种遗传性肾脏病。该病是由编码Ⅳ型胶原蛋白α3、α4、α5链的COL4A3、COL4A4和COL4A5基因突变导致。
X 连锁Alport综合征(XLAS);
常染色体隐性 Alport 综合征(ARAS);
常染色体显性 Alport 综合征(ADAS);
双基因Alport综合征(digenic Alport syndrome)。
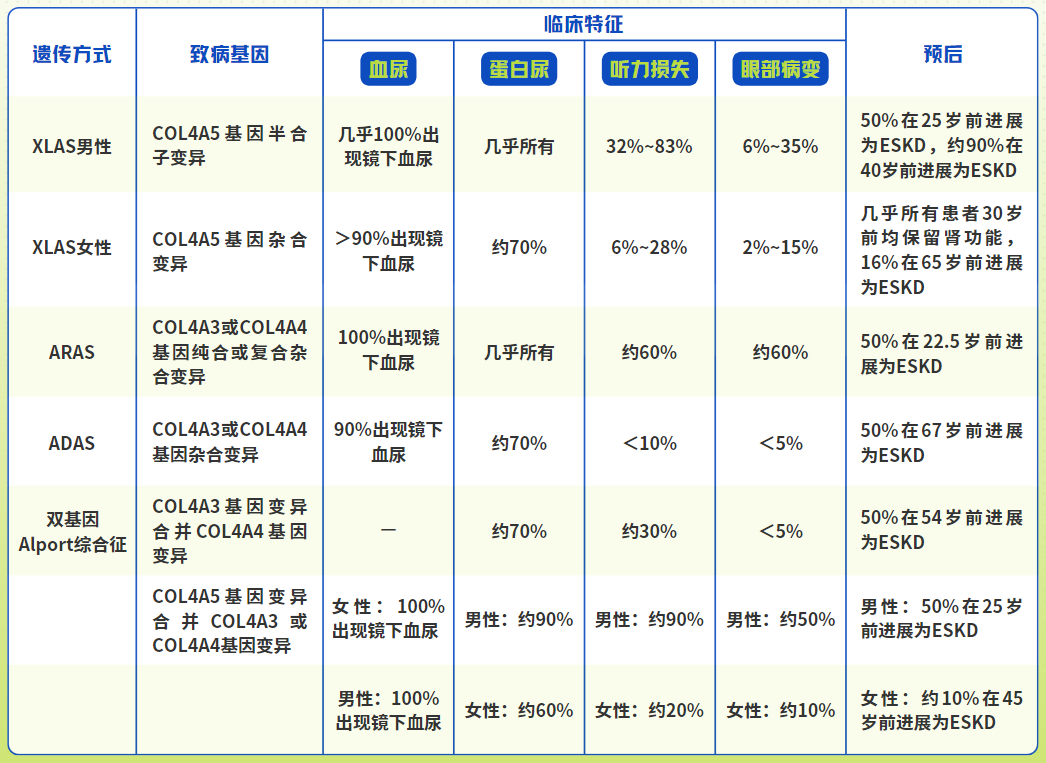
图1 Alport综合征不同遗传方式的致病基因、临床特征及预后情况,其中ESKD为终末肾脏病, “ 一 ” 表示目前缺乏相关研究数据。
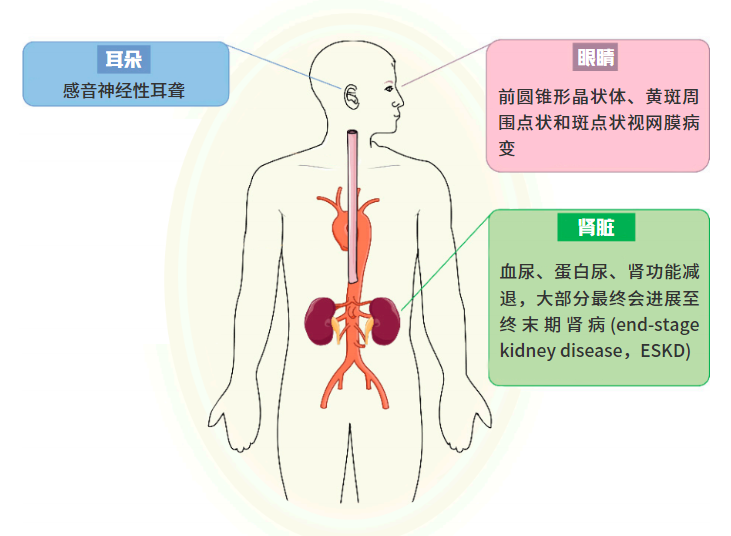
孤立性镜下血尿是AS患者早期的表现,AS患者随着年龄增大,均有较高风险出现血尿伴蛋白尿、进行性肾功能减退,甚至合并感音神经性耳聋、眼部异常及平滑肌瘤等肾外表现。
图2 Alport综合征的主要表型特征
Alport综合征诊断依靠临床表现、组织病理、家系分析及基因诊断。其中,基因检测在Alport综合征的确诊、遗传分型、预测疾病进展、指导治疗及产前基因诊断等方面发挥着重要作用,是诊断该疾病的金标准。
图3 Alport综合征的诊断流程
临床主要表现为持续性肾小球源性血尿或血尿伴蛋白尿的患者,具有以下任一项即可诊断为Alport综合征:
肾组织电镜示GBM致密层撕裂、分层、薄厚不均或篮网状改变。
肾脏组织基底膜Ⅳ型胶原α3、α4、α5链免疫荧光染色异常或EBM Ⅳ型胶原α5链免疫荧光染色异常。
基因检测示COL4A3、COL4A4或COL4A5基因具有致病性变异(包括 ACMG 分级的可能致病性变异)。
Alport综合征尚无根治或病因性治疗措施。目前治疗主要是通过早期药物治疗减缓疾病的进展和进行肾脏替代治疗。
肾素-血管紧张素系统(RAAS)抑制剂,可以通过抑制RAAS活化、调整球管反馈,降低肾小球高滤过而减少蛋白尿,延缓肾小球硬化和疾病进展。
对于进展到终末期肾病的患者需进行肾脏替代治疗,可选择血液透析、腹膜透析和肾脏移植。
助听器有助于改善下降的听力,通过扩大外周声音而减小耳鸣干扰,但不能完全纠正听力异常。
圆锥晶状体或白内障造成的严重视力损害不能通过眼镜或隐形眼镜矫正,晶状体摘除及眼内晶体植入是行之有效的标准治疗。
建议在儿科或成人肾脏内科、耳鼻喉科及眼科规律随诊,避免肾毒性药物、耳毒性药物,避免长期暴露于高噪音环境。
对有生育需求的Alport综合征患者或家系成员,建议在怀孕前进行基因检测,确定基因突变的位点,并进行合理的遗传咨询和生育指导。同时,已孕妈妈建议做产前基因诊断,以实现优生优育。








 粤公网安备44030502000004号
粤公网安备44030502000004号




