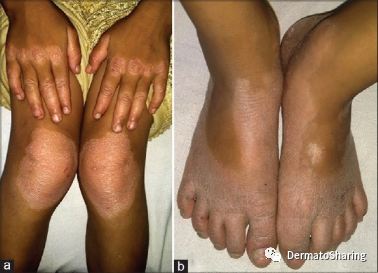
进行性对称性红斑角皮症(Progressive Symmetric Erythrokeratoderma,PSEK),亦称Gottron’s综合征、Gottron’s病、Darier-Gottron综合征,是一种罕见遗传性皮肤病。
该病最初于1911年由Darier首次描述并命名为“Erythrokeratodermie Verruqueuse en Nappes, Symmetrique et Progressive”,后于1922年由Gottron命名为PSEK。
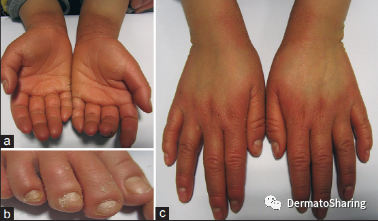
红斑角皮症(erythrokeatodermias)是一组罕见遗传性角化性疾病,以不同程度的红斑及角化过度为典型特征,分为非综合征型(包括2种主要亚型)与综合征型(如KID综合征、HID综合征)。其中2种主要非综合征亚型即为PSEK与可变性红斑角化症(erythrokeratoderma variabilis, EKV)。
1 病因学
多数病例呈常染色体显性遗传,外显率不完全,表达率可变。
具体致病基因尚未知,但多数病例均可见兜甲蛋白编码基因突变。亦有连接蛋白、KDSR((3-ketodihydrosphingosine reductase,编码1种参与神经酰胺合成通路的关键酶,隐性遗传)、KRT83(功能丢失性突变,隐性遗传)、TYR基因突变报道。
兜甲蛋白是角化胞膜的主要结构组分。
连接蛋白是皮肤、神经系统、内耳、角膜及晶状体中缝隙连接的主要蛋白亚单位。

2 流行病学
目前世界范围所报道病例<100例,其中约50%伴阳性家族史,其余多属于自发突变所致。
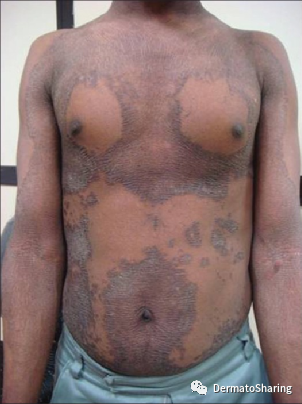
3 临床表现
典型PSEK表现为界清、红色、角化过度性斑块,伴显著的外周红斑,大致对称分布于头面部、四肢(伸侧多见;约50%可见掌跖角化;腕、踝部亦可受累)、肩带区、臀部、腹股沟。皮损呈非迁徙性。
儿童早期(10岁前)发病,进展数年,青春期后形态、色调、位置保持稳定。罕见情况下,青春期后可部分缓解。亦有晚发型报道。
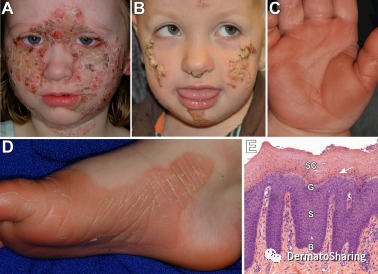
(A、B、C、D,皮损表现;E,显著的棘层增厚、颗粒层透明角质颗粒减少或缺失、角质层细胞核存留)
患者心理、体力常无影响。
PSEK具有显著的遗传异质性,临床表现在家庭内及家庭间具有巨大变异性。
PSEK与 高腭穹、舌裂、漏斗胸、对称性并指症、毛囊角化病、皮脂腺白内障、智力迟钝、智力低下、嗜睡、肾病综合征、惊厥等相关。
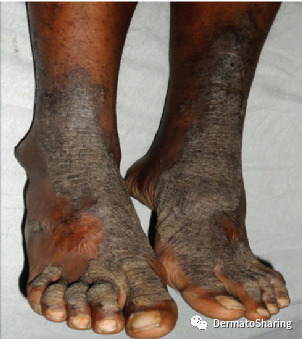
有皮肤癣菌病、假性阿洪病、肾病综合征、点状掌跖角皮症等伴发报道。
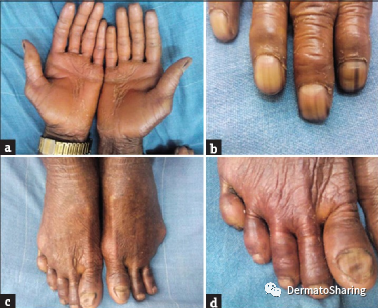
(a,掌部角化;b,纵向黑甲;c,左足趾自截;d,右足趾假性阿洪病)
4 组织病理
显著正角化过度、局灶性角化不全、棘层增厚、教堂塔尖样表现、颗粒层正常或增厚,真皮浅层血管周淋巴细胞浸润。
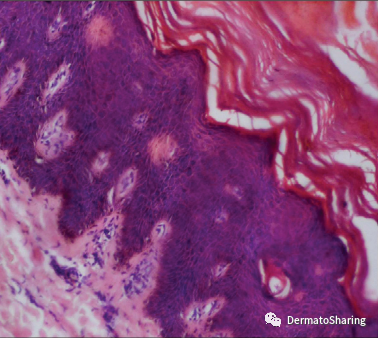
5诊断
依据临床表现多可直接诊断。
组织病理缺乏特异性。
基因筛查可辅助确诊。
鉴别诊断主要包括EKV、银屑病、毛发红糠疹等。
|
表1 PSEK与EKV鉴别点 |
||
|
特点 |
PSEK |
EKV |
|
发病年龄 |
婴儿期或儿童期 |
出生时或婴儿期 |
|
基因突变 |
尚未确认 |
GJB3基因、GJB4基因 |
|
进展 |
皮损在受累区缓慢延伸直至青春期 |
皮损不进展,但会迁移 |
|
皮损豁免区 |
躯干 |
面部 |
|
皮损特点 |
非游走性、界清红斑、角化过度性斑块伴外周红斑 |
固定角化过度性斑块,游走性/一过性红色鳞屑性斑块,无外周红斑 |
|
皮损分布 |
对称性 |
非对称 |
|
加重因素 |
无 |
应激(情绪、生理)、温度变化 |
|
掌跖受累 |
更常见 |
更少见 |
6治疗
口服维A酸类药物(异维A酸)、外用维A酸类、外用激素、角质溶解剂、激素、卡泊三醇均有应用报道。
参考文献
[1] Duan, Y, L Li, Y He, et al., Analysis of TYR Gene Pathogenic Variants in a Chinese Mongolian Family with Progressive Symmetric Erythrokeratoderma[J]. Indian Dermatol Online J, 2021. 12(6): 896-899.DOI: 10.4103/idoj.IDOJ_665_20.
[2] Tiwary, AK and P Kumar, Progressive Symmetrical Erythrokeratoderma Associated with Punctate Palmoplantarkeratoderma[J]. Indian Dermatol Online J, 2019. 10(2): 183-186.DOI: 10.4103/idoj.IDOJ_105_18.
[3] Shah, K, M Ansar, ZU Mughal, et al., Recessive progressive symmetric erythrokeratoderma results from a homozygous loss-of-function mutation of KRT83 and is allelic with dominant monilethrix[J]. J Med Genet, 2017. 54(3): 186-189.DOI: 10.1136/jmedgenet-2016-104107.
[4] Boyden, LM, NG Vincent, J Zhou, et al., Mutations in KDSR Cause Recessive Progressive Symmetric Erythrokeratoderma[J]. Am J Hum Genet, 2017. 100(6): 978-984.DOI: 10.1016/j.ajhg.2017.05.003.
[5] Asha, GS, DV Lakshmi, K Shilpa, et al., Late Onset Progressive Symmetric Erythrokeratoderma with Pseudo Ainhum[J]. Indian J Dermatol, 2016. 61(4): 448-50.DOI: 10.4103/0019-5154.185727.
[6] Wu, F, J Chen, ZH Li, et al., Progressive symmetric erythrokeratoderma with dermatophytosis[J]. Indian J Dermatol Venereol Leprol, 2014. 80(4): 345-7.DOI: 10.4103/0378-6323.136917.
[7] Gupta, LK, P Saini, AK Khare, et al., Progressive symmetric erythrokeratoderma: report of an Indian family[J]. Int J Dermatol, 2014. 53(5): e317-9.DOI: 10.1111/ijd.12334.
[8] Chander, R, M Jabeen, M Barara, et al., Progressive symmetric erythrokeratoderma with unusual associations[J]. Indian J Dermatol, 2014. 59(3): 317.DOI: 10.4103/0019-5154.131476.
[9] Sacchidanand, S, MS Sahana, SG Kamoji, et al., Progressive symmetric erythrokeratoderma with nephrotic syndrome: Coincidence or new association?[J]. Indian Dermatol Online J, 2013. 4(4): 347-9.DOI: 10.4103/2229-5178.120680.
[10] Prabhu, S, SD Shenoi, SB Pai, et al., Progressive and symmetric erythrokeratoderma of adult onset: A rare case[J]. Indian Dermatol Online J, 2010. 1(1): 43-5.DOI: 10.4103/2229-5178.73261.







 粤公网安备44030502000004号
粤公网安备44030502000004号




